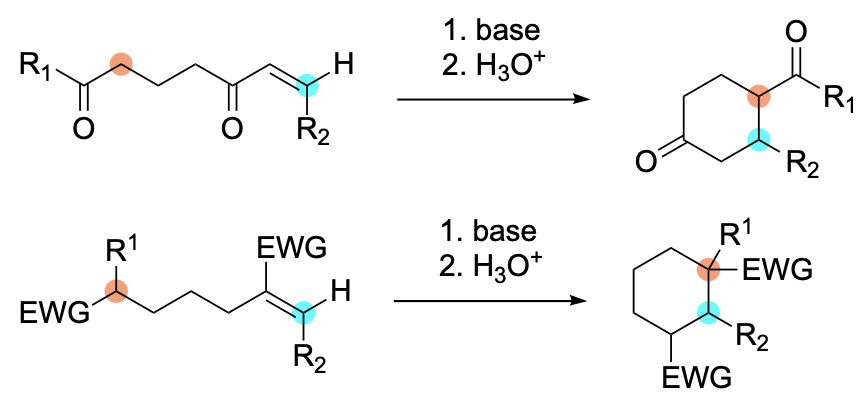
The Intramolecular Michael Reaction
Abstract
 Graphical Abstract by Rebecca Anderson
Graphical Abstract by Rebecca Anderson
Like its intermolecular counterpart, the intramolecular Michael reaction involves the addition of a nucleophile, often referred to as the donor, to an acceptor, usually an olefin bearing one or more functional groups capable of stabilizing a carbanion.
Strictly speaking, the term Michael reaction refers to the 1,4-, or conjugate, addition of a carbanion to an acceptor under basic conditions. However, just as the scope of a previous review dealing with the Michael reaction was expanded to encompass a wide range of acceptors and noncarbon-centered donors, so too has the scope of this chapter. Furthermore, reactions occurring under both basic and acidic conditions are discussed.
As suggested by the name, the intramolecular Michael reaction most often leads to the formation of a ring, either carbo- or heterocyclic. However, there are many examples of sequential, or tandem, Michael reactions wherein the first inter- or intramolecular Michael reaction is followed directly by an intramolecular variant leading to the formation of more than one ring. In other instances, a nucleophile is delivered to the β-carbon atom of an acceptor in a process which does not lead to the production of a ring. These transformations are referred to as heteroconjugate addition reactions.
In contrast to the intermolecular counterpart, the intramolecular Michael reaction has been formally reviewed only once. Two contemporary publications, however, have focused on The Reaction">the reaction and serve to highlight recent developments. The first example dates back to 1898 when Guthzeit reported that on standing at room temperature for a year, or upon treatment with a “small amount” of pyridine, ethyl α-carboxyglutaconate transforms to the “bimeric” ester. Subsequent reinvestigation by Ingold and co-workers led to the revised structure and to the suggestion that it arose via an initial inter- followed by an intramolecular Michael reaction.
The first systematic study of the intramolecular Michael reaction appeared in 1945. Thus, treatment of a variety of substituted ethyl coumarinates with sodium alkoxide in alcohol solvent leads to coumarins. Of particular interest is the fact that the reaction proceeds efficiently even when the β carbon of the acceptor is substituted with two sterically demanding groups. This apparent lack of retardation by double substitution at the β carbon contrasts sharply with the intermolecular Michael reaction and appears to be general. Another example highlighting the susceptibility of sterically hindered systems to participate in intra- but not intermolecular Michael reactions comes from studies concerning the mode of activation of the antibiotics calicheamicin and esperamicin.
Access to the tetracyclic precursor of a taxol analog was achieved via an intramolecular Michael addition to the relatively hindered β carbon of enone.
The intramolecular Michael reaction again formed the subject of publications appearing in 1951, 1957, and 1962. During the 1960s, comparatively little study or use of the reaction was reported. A notable and historically significant exception, however, was Corey’s use of it in a total synthesis of longifolene.
This approach to the construction of tricyclic systems is similar to that suggested mechanistically in the base-induced conversion of santonin to santonic acid.
The 1970s and 1980s witnessed a dramatic increase in uses of the intramolecular Michael reaction. Much of the chemistry discussed in this chapter focuses upon material discovered during this time period.